All test results that fall outside the specifications or acceptance criteria established in drug applications, drug master files (DMFs), official compendia, or by the manufacturer including all in-process laboratory tests that are outside of established specifications.
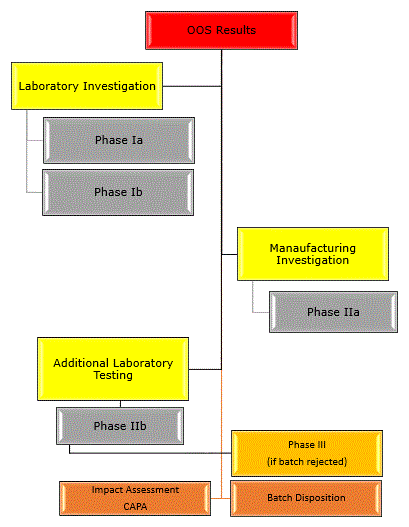
Regulations require that an investigation to determine the cause of OOS results.
The source of OOS results should be identified either as an aberration of measurement process (Laboratory analysis) or an aberration of manufacturing process.
Obviously investigation starts from laboratory as OOS results generates from laboratory during testing. Investigation contains different phases as detailed below.
Analysts should check the data for compliance with test specifications before discarding test preparations or standard preparations. When unexpected results are obtained and no obvious explanation exists, test preparations should be retained, if stable, and the analyst should inform the supervisor for initiation of OOS investigation
Phase-1a Investigation:
The main purpose of Phase 1a investigation is to verify whether the generated OOS results are accurate, and the testing was performed without any deviation to respective testing procedure and practices.
Focus to be given to ensure no obvious errors occurred during the testing process.
Examples of obvious errors.
- Incorrect documents
- Incorrect samples, standard
- Incorrect or dirty glassware
- Calculation errors
- Power outage
- Equipment failure or Incorrect Instrument Parameters
- Preparation errors
- Non-qualified instruments or untrained analysts.
- Issues with environment temperature & Humidity
- Incorrect media or reagents, incorrect incubation times
The following steps should be taken as part of the Laboratory assessment:
- Discuss the test method with the analyst; confirm analyst knowledge of and performance of the correct procedure.
- Examine the raw data obtained in the analysis, including chromatograms and spectra, and identify anomalous or suspect information.
- Verify that the calculations used to convert raw data values into a final test result are scientifically sound, appropriate, and correct; also determine if unauthorized or unvalidated changes have been made to automated calculation methods.
- Confirm the performance of the instruments.
- Determine that appropriate reference standards, solvents, reagents, and other solutions were used and that they met quality control specifications.
- Evaluate the performance of the test method to ensure that it is performing according to the standard expected based on method validation data and historical data.
- Fully document and preserve records of this laboratory assessment.
- Review the history and evaluate the previous trends.
A basic hypothesis testing (Investigative analysis) with retained sample preparations can also be considered as part of Phase 1a investigation to identify what might have happened during analysis.
- Same vials can be re-injected, same vial re-injections can provide strong evidence that problem should be attributed instrument performance.
- Same solutions can be re-injected, reinjections of same solutions can provide strong evidence that the problem should be attributed to the instrument or vials, rather than the sample or its preparation.
- Solutions can be re-diluted from stock and re-injected, re-diluted re-injections can provide strong evidence that the problem should be attributed the sample or its preparation.
- Investigation shall be extended to Phase Ib, when the phase Ia investigations did not reveal an assignable laboratory error.
Phase-1b Investigation:
Phase 1b consists of detailed hypothesis testing (or) investigative analysis. i.e testing performed to help confirm or discount a possible root cause i.e what might have happened that can be tested:- Multiple hypothesis can be explored based on the suspection of Phase 1a investigation.
it may include but not limited to
- Testing regarding sample filtration, sonication /extraction to evaluate the impact on sample results and possibility of improper filtration, sonication/extraction
- Testing of spiked samples to confirm or discount the possibility of contamination.
- Examination of the original dosage unit to determine whether it was damaged during laboratory handling in a way that affected its performance for release rate testing of certain specialized dosage form drugs.
- Testing of retention/control samples or additional stability samples.
- Simulations with different preparations, different columns and different conditions based on suspection.
Phase 1b investigations should be driven by written and approved instructions against hypothesis. Description of the testing should be written, and then approved by QA/Contract Giver/QA equivalent prior to initiating investigative testing.
The requirements of investigative testing listed below:
The description must fully document
- The hypothesis to the test the root cause being investigated.
- What samples will be tested.
- The exact execution of the testing.
- How the data will be evaluated
Investigative testing may not be used to replace an original suspect analytical results. It may only be used to confirm or discount a probable cause.
In summary, when clear evidence of laboratory error exists, laboratory testing results should be invalidated. When evidence of laboratory error remains unclear, investigation should be extended to manufacturing firm to determine what caused the unexpected results. It should not be assumed that OOS test results are attributable to analytical error without performing and documenting an investigation. Both the initial laboratory assessment and the following OOS investigation should be documented fully.
A full-scale investigation should include a review of production and sampling procedures, and will often include additional laboratory testing
Phase IIa Investigation:
Phase IIa investigation consists of manufacturing investigation. Where all cross-functional departments like production, warehouse, vendors etc shall be communicated regarding OOS & laboratory investigation details to initiate investigation at their end as applicable.
Relevant functions shall review the following (but not limited to) to identify the, if any assignable cause
- Manufacturing process sequence and related critical parameters
- Review of executed manufacturing records
- Review of production activity to confirm that operator is properly trained and appropriate standard procedure, batch records are available and followed
- Review of calculations and computer generated data
- Review of performance of machine(s)/personnel(s) associated with the batch.
- Evaluation of storage & transportation of raw materials and samples used in the batch and nature of materials used in the batch.
- Evolution of sampling procedure
- Review of historical trends where applicable
- Impact on earlier supplies
- Vendor investigation report review where necessary
If this part of the OOS investigation confirms the OOS result and is successful in identifying its root cause, the OOS investigation may be terminated and the product shall be rejected.
Phase IIb Investigation:
Additional Laboratory testing involves retesting of the sample.
Retesting can be done from a portion of original sample. The sample used for the retesting should be taken from the same homogeneous material that was originally collected from the lot, tested, and yielded the OOS results. For a liquid, it may be from the original unit liquid product or composite of the liquid product; for a solid, it may be an additional weighing from the same sample composite prepared for the original test.
- If insufficient quantity of the original sample remains to perform all further testing then the procedure for obtaining a re-sample must be discussed and agreed by QA/Contract Giver/QA equivalent. The process of obtaining the re-sample should be recorded within the laboratory investigation.
- The decision to retest should be based on sound scientific judgement. The test plan must be approved before re testing occurs.
If original sample quantity is insufficient or investigation reveals sampling error then re-sampling shall be performed by the same qualified, validated methods that are used for initial sample collection.
The maximum number of retests to be performed on a sample should be specified in advance in a written standard operating procedure (SOP). The number may vary depending upon the variability of the particular test method employed, but should be based on scientifically sound principles.
The number of retests should not be adjusted depending on the results obtained. The firm's predetermined retesting procedures should contain a point at which the additional testing ends and the batch is evaluated. If the results are unsatisfactory at this point, the batch is suspect and must be rejected or held pending further investigation
After retesting,
OOS Confirmed:
If OOS results are confirmed then batch shall be rejected by reporting initial OOS results.
The investigation changes from an OOS investigation into a batch failure investigation, which must be extended to other batches or products that may have been associated with the specific failure as part of Phase III.
OOS Conclusive:
If retesting results found with in specification and assignable cause identified then report the retest results and invalidate the initial OOS results. CAPA shall be proposed accordingly and impact assessment shall be done.
OOS Inconclusive:
If retesting results found with in specification but no assignable cause identified then report all the results and decide on batch disposition.
All test results, both passing and suspect, should be reported (in all QC documents and any Certificates of Analysis) and all data has to be considered in batch release decisions
For inconclusive investigations — in cases where an investigation:-
(1) does not reveal a cause for the OOS test result and
(2) Does not confirm the OOS result
the OOS result should be given full consideration (most probable cause determined) in the batch or lot disposition decision by the certifying QP and the potential for a batch specific variation also needs considering.
Any decision to release a batch, in spite of an initial OOS result that has not been invalidated, should come only after a full investigation has shown that the OOS result does not reflect the quality of the batch. In making such a decision, Quality Assurance/QP should always err on the side of caution.
The QA might still ultimately decide to release the batch. For example, a firm might consider release of the product under the following scenario:
A product has an acceptable composite assay range of 90.0 to 110.0 percent. The initial (OOS) assay result is 89.5 percent. Subsequent sample preparations from the original sample yield the following retest results: 99.0, 98.9, 99.0, 99.1, 98.8, 99.1, and 99.0 percent.
A comprehensive laboratory investigation (Phase 1) fails to reveal any laboratory error.
Review of events during production of the batch reveals no aberrations or indication of unusual process variation. Review of the manufacturing process and product history demonstrates that the process is robust.
The passing retest results are all well within the known limits of variability of the method used. Batch results from in-process monitoring, content uniformity, dissolution, and other tests are consistent with the passing retest results.
After a thorough investigation, a firm’s QA might conclude that the initial OOS result did not reflect the true quality of the batch.
It is noteworthy in this scenario that the original, thorough laboratory investigation failed to find any assignable cause. However, if subsequent investigation nonetheless concludes that the source of the OOS result was a cause unrelated to the manufacturing process, in response to this atypical failure to detect the laboratory deviation, it is essential that the investigation include appropriate follow-up and scrutiny to prevent recurrence of the laboratory error(s) that could have led to the OOS result.
As the above example illustrates, any decision to release a batch, in spite of an initial OOS result that has not been invalidated, should come only after a full investigation has shown that the OOS result does not reflect the quality of the batch.
it was very helpful. thank you so much for such well explained article.
ReplyDelete